What does a peak in ChIP-seq tell you?
What does a peak in ChIP-seq tell you?
Peak calling is a computational method used to identify areas in a genome that have been enriched with aligned reads as a consequence of performing a ChIP-sequencing or MeDIP-seq experiment. These areas are those where a protein interacts with DNA.
What is peak calling and why is it needed for ChIP-seq analysis?
Peak calling is one of the first steps in the analysis of these data. Peak calling consists of two sub-problems: identifying candidate peaks and testing candidate peaks for statistical significance. We surveyed 30 methods and identified 12 features of the two sub-problems that distinguish methods from each other.
What does ATAC seq measure?
The assay for transposase-accessible chromatin with sequencing (ATAC-Seq) is a popular method for determining chromatin accessibility across the genome. By sequencing regions of open chromatin, ATAC-Seq can help you uncover how chromatin packaging and other factors affect gene expression.
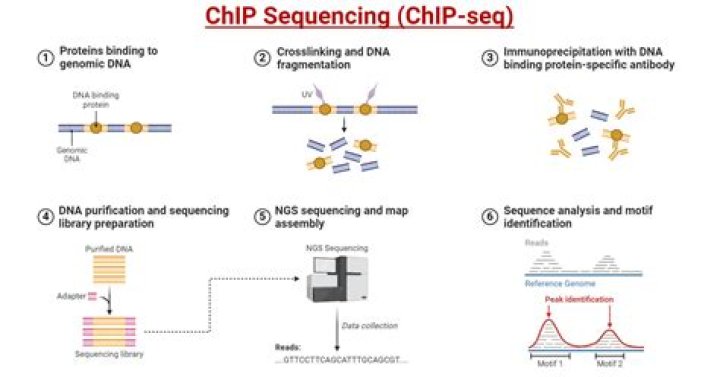
What is ChIP-seq used for?
ChIP-Seq identifies the binding sites of DNA-associated proteins and can be used to map global binding sites for a given protein. ChIP-Seq typically starts with crosslinking of DNA-protein complexes. Samples are then fragmented and treated with an exonuclease to trim unbound oligonucleotides.
How many peaks does ChIP-seq have?
With ChIP-seq, the alignment of the reads to the genome results in two peaks (one on each strand) that flank the binding location of the protein or nucleosome of interest.
How many reads do you need for ChIP-seq?
What is the minimum number of reads per sample and sequencing format for ChIP-Seq? For studies targeting transcription factors, Illumina recommends 5–15 M 1×35–1×50 reads per sample. For studies targeting histone modifications, we recommend 50–90M 1×35–1×50 reads.
What can you do with ChIP-seq data?
Advanced applications. Because abundant ChIP-seq data are available for several well-studied cell types, it is useful to leverage information from these cell types to infer genome dynamics or to annotate the epigenetic landscape of other cell types with fewer additional experiments.
How expensive is ATAC-seq?
Costs for standard and single-cell ATAC-seq library construction are $350 and $2,150, respectively, for Northwestern users.
What can ATAC-seq do?
Is ChIP-seq expensive?
For high-resolution profiling of an entire large genome, ChIP-Seq is already less expensive than ChIP-chip; but depending on the genome size and the depth of sequencing needed, a ChIP-chip experiment on carefully selected regions using a customized microarray may yield as much biological understanding.
What is MeDiP Seq used for?
MeDIP-seq requires less input DNA, demonstrates solid correlation with array-based methods [7], and allows for broader unbiased genome investigations including unannotated regions and repetitive elements. Owing to these advantages, MeDIP-seq is widely employed for medical and agriculture epigenetics research.
What is differential peak calling in ChIP-seq?
In the context of ChIP-exo, this process is known as ‘peak-pair calling’. Differential peak calling is about identifying significant differences in two ChIP-seq signals.
What is a broad peak in ChIP-seq?
ChIP-seq peaks from epigenomic data can be narrow, broad or gapped. Histone marks such as H3K9me3 or H3K27me3 are broad while others such as H3K4me3 and proteins such as CTCF are narrow. Other DNA binding proteins such as HP1, Lamins (Lamin A or B), HMGA etc. form broad peaks or domains.
What is meant by peak calling in genomics?
Peak calling. Peak calling is a computational method used to identify areas in a genome that have been enriched with aligned reads as a consequence of performing a ChIP-sequencing or MeDIP-seq experiment. These areas are those where a protein interacts with DNA. When the protein is a transcription factor,…
